Reproducible scripting, AlphaSimR, Homework
Jean-Luc Jannink
USDA-ARS / CornellJanuary 31, 2022
Last updated: 2022-02-01
Checks: 7 0
Knit directory: QuantitativeGen/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20220124) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 79038f7. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/Lab2_ProgrammingAlphaSimR.Rmd) and HTML (docs/Lab2_ProgrammingAlphaSimR.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 79038f7 | LucianoRogerio | 2022-02-01 | Homework 2 |
| html | 79038f7 | LucianoRogerio | 2022-02-01 | Homework 2 |
Preliminaries
Learning objectives
- A bit more on reproducible scripting
Some other things that matter to me to ensure communicability and reproducibility
- The main README.md file should contain information about the conditions under which the script was run
- I like the script to have a predictable strucure: load and document the packages used, if there is a stochastic component, set the random seed, set and document the parameter values that will affect the script output
- Use a consistent directory structure. Happily, such a consistent structure is enforced by
workflowr
- Use the package
hereto make file locations relative to the base project folder
AlphaSimR
AlphaSimRis a package to simulate breeding populations and tasks. It is not completely intuitive nor completely well-documented. We will want to use it ultimately to optimize breeding schemes.Prepare a Homework
In class, we have discussed two interesting phenomena related to quantitative traits:
- The “outbreak of variation” that occurs when a heterozygous individual is self-fertilized
- Regression toward the mean between parents and progeny
This Rmarkdown script contains code to illustrate the former. Your homework will be to write a script that illustrates the latter
Ordering of the script
It’s good to have all scripts in the same order with respect to standard tasks
Load packages first
If your script depends on external packages, load them at the beginning. This shows users early on what the script dependencies are.
packages_used <- c("AlphaSimR", "tidyverse", "workflowr", "here")
ip <- installed.packages()
all_packages_installed <- TRUE
for (package in packages_used){
if (!(package %in% ip[,"Package"])){
print(paste("Please install package", package))
all_packages_installed <- FALSE
}
}#END packages_used
if (!all_packages_installed) stop("Need to install packages")
library(tidyverse)── Attaching packages ─────────────────────────────────────── tidyverse 1.3.1 ──✓ ggplot2 3.3.5 ✓ purrr 0.3.4
✓ tibble 3.1.6 ✓ dplyr 1.0.7
✓ tidyr 1.1.4 ✓ stringr 1.4.0
✓ readr 2.1.1 ✓ forcats 0.5.1── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
x dplyr::filter() masks stats::filter()
x dplyr::lag() masks stats::lag()Notice the conflicts report from loading tidyverse. Two packages (dplyr and stats) both have a function called filter. Since dplyr was loaded after stats, if you use the function filter, it will go to the dplyr version. It is dangerous to rely on what order packages have been loaded to determine which filter function you get. R syntax to prevent ambiguity is to write either dplyr::filter or stats::filter. Using that syntax will make your code more reproducible.
Set file locations relative to the project folder using here
here::i_am("analysis/Lab2_ProgrammingAlphaSimR.Rmd")There are two functions I use in the here package: here::i_am and here::here. here::i_am helps here “understand” the folder structure. You give it the name of the script that is currently running, and the path to that script from the base of the overall analysis folder. From that, here deduces where the base of the folder is. After that, here::here locates files from that folder base. If you use the here package, you can send anyone a complicated zipped folder, and they should be able to run the analysis out of the box, regardless of where they put the folder in their own file system.
Document packages used
This chunk creates a “README.md” file that documents the packages and versions used for future reference.
source(here::here("code/addToREADME.R"))
addToREADME(paste0("## ", rmarkdown::metadata$title), append=F)
addToREADME(c(date(), ""))
packages_info <- ip[packages_used, c("Package", "Version", "Built")]
addToREADME(c("The packages used in this script are:", "Package, Version, Built"))
apply(packages_info, 1, function(vec) addToREADME(paste(vec, collapse=" "))) AlphaSimR tidyverse workflowr
"AlphaSimR 1.0.4 4.1.1" "tidyverse 1.3.1 4.1.0" "workflowr 1.7.0 4.1.1"
here
"here 1.0.1 4.1.0" addToREADME("")Hyperlink the README file to the report. That makes it easy to find.
Set random seed
AlphaSimR generates many random numbers (e.g., to simulate Mendelian random segregation). If you want the result of the analysis to come out the same each time (there are pros and cons) you need to set the random seed. Note that workflowr does this by default. If you are not using that package, then be explicit.
random_seed <- 45678
set.seed(random_seed)
addToREADME(c(paste("The random seed is", random_seed), ""))Script parameters
If the behavior of your script depends on parameters that you set, initialize them early on.
nFounders <- 100
nChr <- 10 # Number of chromosomes
nSitesPerChr <- 1000 # Number of segregating sites _per chromosome_
nQTLperChr <- 10 # Vary this parameter to get oligo- versus poly- genic traits
nF1s <- 200 # We are going to make F1s to test outbreak of variation
nF2s <- 200 # We are going to make F2s to test outbreak of variationParameters to README
It’s good to have all the information together in the README. Chunks of code like this do not need to be included in the report. To exclude them use the “include=FALSE” option in the chunk.
Simulating some classical results
This script uses AlphaSimR to simulate the “outbreak of variation” that arises when you self-fertilize a hybrid.
AlphaSimR populations
The basic object of AlphaSimR is the population. To make founders, you first make founder haplotypes from a coalescent simulation, then you define simulation parameters that will link their genetic variation to phenotypic variation, then you make a first diploid population from the founder haplotypes.
# Create haplotypes for founder population of outbred individuals
# Note: default effective population size for runMacs is 100
founderHaps <- AlphaSimR::runMacs(nInd=nFounders, nChr=nChr,
segSites=nSitesPerChr)
# founderHaps <- AlphaSimR::quickHaplo(nInd=nFounders, nChr=nChr,
# segSites=nSitesPerChr)
# New global simulation parameters from founder haplotypes
SP <- AlphaSimR::SimParam$new(founderHaps)
# Additive trait architecture
# By default, the genetic variance will be 1
SP$addTraitA(nQtlPerChr=nQTLperChr)
# Create a new population of founders
founders <- AlphaSimR::newPop(founderHaps, simParam=SP)
str(founders)Formal class 'Pop' [package "AlphaSimR"] with 18 slots
..@ id : chr [1:100] "1" "2" "3" "4" ...
..@ iid : int [1:100] 1 2 3 4 5 6 7 8 9 10 ...
..@ mother : chr [1:100] "0" "0" "0" "0" ...
..@ father : chr [1:100] "0" "0" "0" "0" ...
..@ sex : chr [1:100] "H" "H" "H" "H" ...
..@ nTraits: int 1
..@ gv : num [1:100, 1] 0.267 0.158 2.274 -0.655 1.145 ...
..@ pheno : num [1:100, 1] NA NA NA NA NA NA NA NA NA NA ...
..@ ebv : num[1:100, 0 ]
..@ gxe :List of 1
.. ..$ : NULL
..@ fixEff : int [1:100] 1 1 1 1 1 1 1 1 1 1 ...
..@ reps : num [1:100] 1 1 1 1 1 1 1 1 1 1 ...
..@ misc :List of 100
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. ..$ : NULL
.. .. [list output truncated]
..@ nInd : int 100
..@ nChr : int 10
..@ ploidy : int 2
..@ nLoci : int [1:10] 1000 1000 1000 1000 1000 1000 1000 1000 1000 1000
..@ geno :List of 10
.. ..$ : raw [1:125, 1:2, 1:100] f6 aa ea 90 ...
.. ..$ : raw [1:125, 1:2, 1:100] 20 f0 58 ba ...
.. ..$ : raw [1:125, 1:2, 1:100] 01 28 d4 2c ...
.. ..$ : raw [1:125, 1:2, 1:100] 27 20 65 71 ...
.. ..$ : raw [1:125, 1:2, 1:100] 25 1d 17 a5 ...
.. ..$ : raw [1:125, 1:2, 1:100] ed 70 06 00 ...
.. ..$ : raw [1:125, 1:2, 1:100] 00 82 2b 0d ...
.. ..$ : raw [1:125, 1:2, 1:100] 13 4d 88 75 ...
.. ..$ : raw [1:125, 1:2, 1:100] 74 28 51 00 ...
.. ..$ : raw [1:125, 1:2, 1:100] 16 89 84 a9 ...
.. ..- attr(*, "dim")= int [1:2] 10 1Population information
The population has ids. The @mother and @father ids are all zero because this population was made from founder haplotypes, and so does not have diploid parents. The genotypic values gv of the population are calculated for the trait created using SP$addTraitA(nQtlPerChr=nQTLperChr). Given that there are 10 chromosomes and 10 QTL per chromosome, there are 3^(nChr*nQTLperChr) = 5.1537752^{47} different possible genotypic values. The realized genotypic values are accessible with the function gv(founders)
From here, you can treat this population like a named vector using the square braces extraction operator [ ]. Extract individuals by their @id or just by their order in the population using an integer index. For example, pick three random individuals from a population and list their ids. Pick the one with the first id in alphabetical order.
test <- founders[c(2, 3, 5, 7, 11)]
testID <- test@id
alphaInd <- test[testID %>% order %>% .[1]] # Put testID in alphabetical order
print(testID)[1] "2" "3" "5" "7" "11"print(alphaInd)An object of class "Pop"
Ploidy: 2
Individuals: 1
Chromosomes: 10
Loci: 10000
Traits: 1 print(alphaInd@id)[1] "11"Outbreak of variation
Emerson and East (1913) showed that if you crossed two inbreds, the hybrid had similar variation to each inbred, but if you then selfed the hybrid, the offspring varied substantially. This code simulates that result. First, self the founders to homozygosity. The function self self-fertilizes individuals from the population. By default, it creates one selfed individual per parent (controllable with the parameter nProgeny), so this works nicely for single-seed descent.
# Self-pollinate to for a few generations
nGenSelf <- 3
inbredPop <- founders
for (gen in 1:nGenSelf){
inbredPop <- AlphaSimR::self(inbredPop)
}Check homozygosity
Just a sanity check that this has, in fact, created a population of 100 individuals that are appropriately homozygous. Loci are coded 0, 1, 2. So qtl == 1 represents the case were a locus is heterozygous. sum(qtl == 1) counts those cases.
qtl <- AlphaSimR::pullQtlGeno(inbredPop)
if (nrow(qtl) != nFounders) stop("The number of individuals is unexpected")
if (ncol(qtl) != nChr * nQTLperChr) stop("The number of QTL is unexpected")
fracHet <- sum(qtl == 1) / (nFounders * nChr * nQTLperChr)
cat("Expected fraction heterozygous", 1 / 2^nGenSelf, "\n",
"Observed fraction heterozygous", fracHet, "+/-",
round(2*sqrt(fracHet*(1-fracHet)/(nFounders*nChr*nQTLperChr)), 3), "\n")Expected fraction heterozygous 0.125
Observed fraction heterozygous 0.0368 +/- 0.004 What was wrong with my reasoning about the Expected fraction heterozygous?
Simulate outbreak of variation
We will
1. pick a random pair of inbred individuals
2. cross that pair
3. find out the variation in genotypic value among the pair’s progeny
4. pick a random F1 progeny
5. self-fertilize that F1
6. find out the variation in genotypic value among the F1’s progeny
We will assume a trait that has a heritability of 0.5 in the base, non-inbred population. In AlphaSimR, that means genetic and error variances of 1.
Pick random pair to make F1 hybrid
randomPair <- inbredPop[sample(nFounders, 2)]Cross the pair to make population of F1s
The crossPlan is a two-column matrix with as many rows as the number of crosses you want to make, the first column is the @id or the index of the seed parent, and likewise for the pollen parent in the second column. We will make 200 F1s from this random pair, so the matrix has 200 rows. You just want to cross individual 1 with individual 2, so each row contains 1:2.
crossPlan <- matrix(rep(1:2, nF1s), ncol=2, byrow=T)
f1_pop <- AlphaSimR::makeCross(randomPair, crossPlan)Find out the phenotypic variation among F1s
When you first make a population, AlphaSimR does not assume it has been phenotyped. You can phenotype it using the setPheno function. Note that if you use setPheno on the same population a second time, that will overwrite the phenotypes from the first time. The genotypic variance can be retrieved using the function varG. Really, varG gives all we need to know, but of course that variance is generally not observable in non-simulated reality.
f1_pop <- AlphaSimR::setPheno(f1_pop, varE=1)
cat("Genotypic variance among F1s", round(AlphaSimR::varG(f1_pop), 3), "\n")Genotypic variance among F1s 0.075 cat("Phenotypic variance among F1s", round(AlphaSimR::varP(f1_pop), 3), "\n")Phenotypic variance among F1s 1.012 hist(AlphaSimR::pheno(f1_pop), main="Histogram of F1 Phenotypes")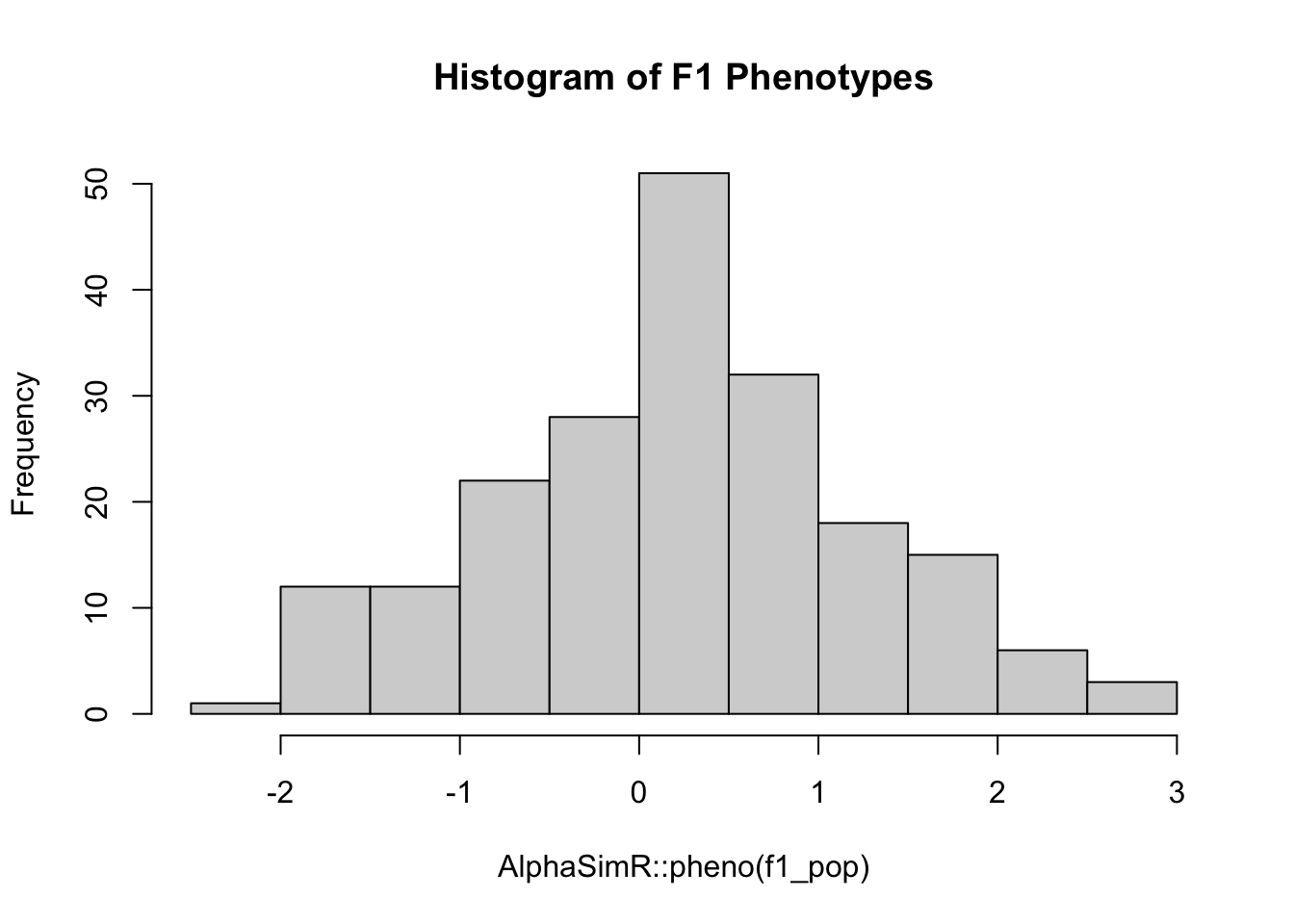
| Version | Author | Date |
|---|---|---|
| 79038f7 | LucianoRogerio | 2022-02-01 |
Pick random F1
randomF1 <- f1_pop[sample(nFounders, 1)]Make F2 and observe phenotypic variance
When you first make a population, AlphaSimR does not assume it has been phenotyped. You can phenotype it using the setPheno function. Note that if you use setPheno on the same population a second time, that will overwrite the phenotypes from the first time. The genotypic variance can be retrieved using the function varG. Really, varG gives all we need to know, but of course that variance is generally not observable in non-simulated reality.
f2_pop <- AlphaSimR::self(randomF1, nProgeny=nF2s)
f2_pop <- AlphaSimR::setPheno(f2_pop, varE=1)
cat("Genotypic variance among F2s", round(AlphaSimR::varG(f2_pop), 3), "\n")Genotypic variance among F2s 0.47 cat("Phenotypic variance among F2s", round(AlphaSimR::varP(f2_pop), 3), "\n")Phenotypic variance among F2s 1.273 hist(AlphaSimR::pheno(f2_pop), main="Histogram of F2 Phenotypes")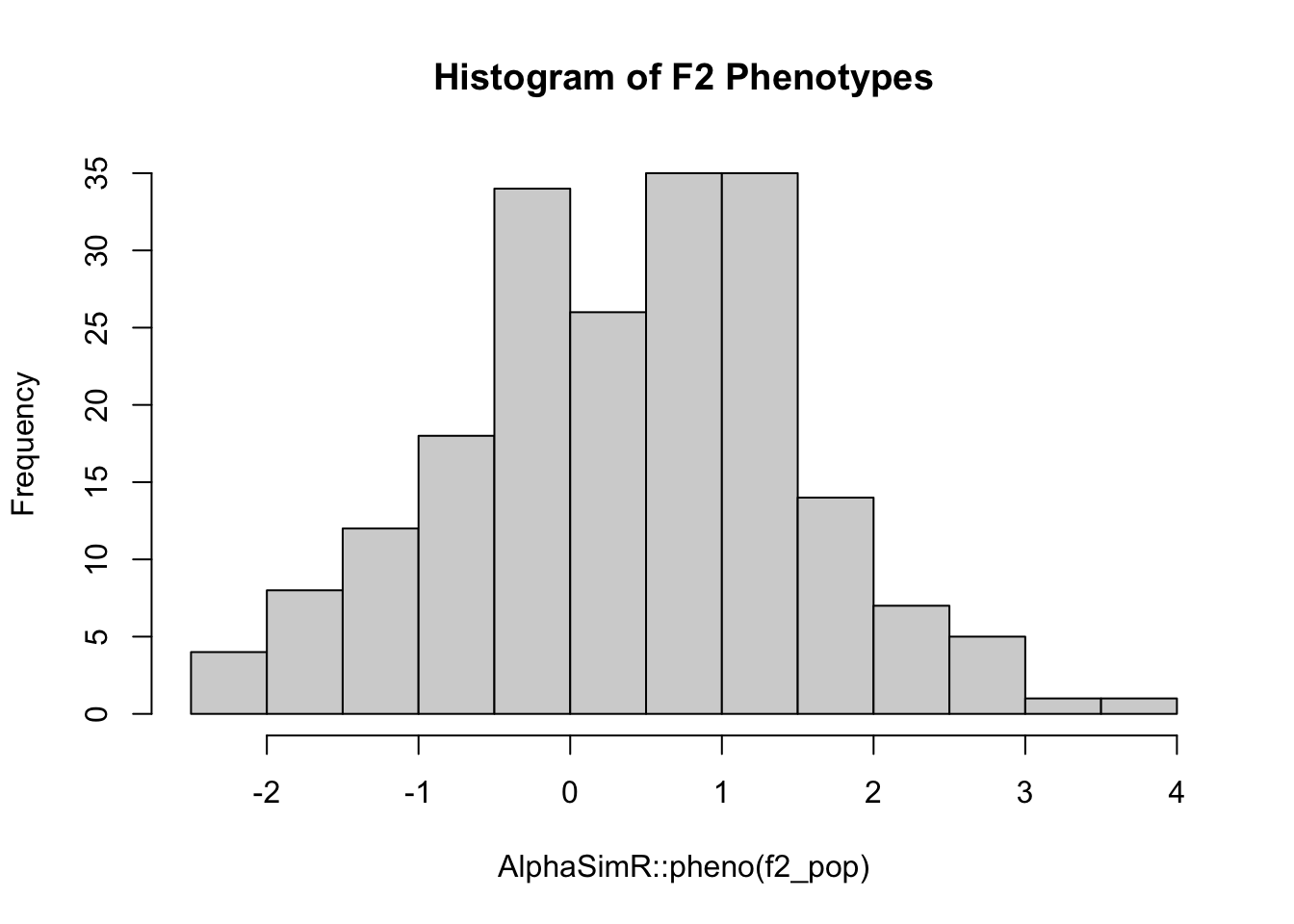
| Version | Author | Date |
|---|---|---|
| 79038f7 | LucianoRogerio | 2022-02-01 |
Illustrate here::here by writing a file
Just to practice again using the here package because it makes life easy.
utils::write.csv(qtl, here::here("output", "QTLgenotypes.csv"))Homework
Illustrate regression to the mean between parents and offspring using AlphaSimR
1. You know how to create a population – create a population of parents
2. You know how to get phenotypes from that population
3. You want to randomly mate that population to get progeny that will be regressed to the mean. Create a crossPlan matrix like I did to generate the F1s, except that each row should have randomly-picked parents, rather than 1 and 2 like for the F1s. There is also a command AlphaSimR::randCross. Check out its documentation. If you use that command, you will have to find the pedigree of the progeny using the @mother and @father ids of the progeny population and match those up to the parent population. If you make the crossPlan, then it gives you the seed and pollen parent ids.
4. Having made the progeny population, phenotype it also
5. Use each row of the crossPlan to find the two parents and calculate their phenotypic mean
6. Make a scatterplot of the progeny phenotypes against the parent mean phenotypes
What is the regression coefficient?
How much closer to the mean are offspring, on average, compared to their parents?
Homework grading
I want to receive a zipped folder with your homework in it one week from today, by midnight. If you use the workflowr package to create the folder structure, that’s great. You should have created an Rmarkdown script that will create an html file that I can look through. So:
1. 40 points for just sending in a zipped folder on time
2. 10 points for each successful step from above, with partial credit
3. I will subtract 5 points for every day the homework is late
sessionInfo()R version 4.1.1 (2021-08-10)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Big Sur 11.6.1
Matrix products: default
LAPACK: /Library/Frameworks/R.framework/Versions/4.1-arm64/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] forcats_0.5.1 stringr_1.4.0 dplyr_1.0.7 purrr_0.3.4
[5] readr_2.1.1 tidyr_1.1.4 tibble_3.1.6 ggplot2_3.3.5
[9] tidyverse_1.3.1
loaded via a namespace (and not attached):
[1] Rcpp_1.0.8 here_1.0.1 lubridate_1.8.0 assertthat_0.2.1
[5] rprojroot_2.0.2 digest_0.6.29 utf8_1.2.2 R6_2.5.1
[9] cellranger_1.1.0 backports_1.4.1 reprex_2.0.1 evaluate_0.14
[13] highr_0.9 httr_1.4.2 pillar_1.6.4 rlang_0.4.12
[17] readxl_1.3.1 rstudioapi_0.13 whisker_0.4 jquerylib_0.1.4
[21] rmarkdown_2.11 bit_4.0.4 munsell_0.5.0 broom_0.7.11
[25] compiler_4.1.1 httpuv_1.6.5 modelr_0.1.8 xfun_0.29
[29] pkgconfig_2.0.3 htmltools_0.5.2 tidyselect_1.1.1 workflowr_1.7.0
[33] fansi_1.0.2 crayon_1.4.2 tzdb_0.2.0 dbplyr_2.1.1
[37] withr_2.4.3 later_1.3.0 grid_4.1.1 jsonlite_1.7.3
[41] gtable_0.3.0 lifecycle_1.0.1 DBI_1.1.2 git2r_0.29.0
[45] magrittr_2.0.1 scales_1.1.1 vroom_1.5.7 cli_3.1.0
[49] stringi_1.7.6 fs_1.5.2 promises_1.2.0.1 xml2_1.3.3
[53] bslib_0.3.1 ellipsis_0.3.2 generics_0.1.1 vctrs_0.3.8
[57] tools_4.1.1 bit64_4.0.5 glue_1.6.0 hms_1.1.1
[61] parallel_4.1.1 fastmap_1.1.0 yaml_2.2.1 colorspace_2.0-2
[65] AlphaSimR_1.0.4 rvest_1.0.2 knitr_1.37 haven_2.4.3
[69] sass_0.4.0