GBS and DArT Markers Imputation
LucianoRogerio
2022-05-09
Last updated: 2022-06-02
Checks: 6 1
Knit directory: EMBRAPAImputation2022/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has staged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20220303) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 95ddcaf. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/DArT2018/
Ignored: data/DArT2020/
Ignored: data/DArT2022/
Ignored: data/GBS/
Ignored: output/DArT2022/
Untracked files:
Untracked: DArTCommonMkrs.log
Untracked: MkrsRefandStudyPop.txt
Untracked: analysis/CheckImp.Rmd
Untracked: analysis/Duplicates.Rmd
Untracked: analysis/ImputationEMBRAPA_DCas22_6902.Rmd
Untracked: analysis/PrepareGenData.Rmd
Untracked: code/.DS_Store
Untracked: code/plink/
Untracked: data/AllDArTDuplicates.txt
Untracked: data/CommonMkrsGBSDArT
Untracked: data/CommonMkrsGBSDArT.txt
Untracked: data/CommonMkrsGBSDArTIssues
Untracked: data/DArTClones.csv
Untracked: data/DArTDupDiscordanceIndex.csv
Untracked: data/DArTDuplicates1.txt
Untracked: data/DArTDuplicates2.txt
Untracked: data/DArTDuplicates3.txt
Untracked: data/DArTDuplicates4.txt
Untracked: data/DArTGPInfo.csv
Untracked: data/DArTGPInfo.xlsx
Untracked: data/DArTGPInfo2.csv
Untracked: data/DArTGPInfo2.xlsx
Untracked: data/DArTGenotypingPlates/
Untracked: out.log
Untracked: output/AllChrDCas22_6902_StudyPopimputed.vcf.gz
Untracked: output/AllChrDCas22_6902_StudyPopimputed.vcf.gz.tbi
Untracked: output/AllChrGBSandDArTsitesCommonClones_RefPopImputed.vcf.gz
Untracked: output/AllChrGBSandDArTsitesCommonClones_RefPopImputed.vcf.gz.tbi
Untracked: output/BRTP_Phenotyping2022.txt
Untracked: output/DCas22_6902/
Untracked: output/DCas22_6902RefPopImputed.vcf.gz
Untracked: output/Dados GBS Atualizados.RData
Untracked: output/DadosGBSAtualizados.rds
Untracked: output/DadosGBSAtualizadosRenomeados.rds
Untracked: output/Duplicates/
Untracked: output/Figures/
Untracked: output/GBSDArTPCA.rds
Untracked: output/GmatrixDArTGS.rds
Untracked: output/GmatrixGBSandDArTGS.rds
Untracked: output/RefPop/
Untracked: output/StudyPop/
Unstaged changes:
Modified: .DS_Store
Modified: analysis/DArTImp.Rmd
Modified: analysis/GBS_DArTImp.Rmd
Modified: analysis/_site.yml
Modified: analysis/index.Rmd
Modified: data/.DS_Store
Modified: output/.DS_Store
Modified: output/out.log
Staged changes:
Modified: .DS_Store
New: analysis/DArTImp.Rmd
New: analysis/GBS_DArTImp.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
Verify the Common SNP Markers (Manihot esculenta v6.1) and Clones
library(here)
system(paste0("bcftools concat ",
"--output ",
"tassel_embrapa_newfastq.bial.recode.vcf.gz ",
"--output-type z --threads 18 ",
paste0("tassel_embrapa_newfastq.chr",1:18,
".bial.recode.vcf.gz",
collapse = " ")))GBS data has 3,456 genotyped clones for 400,122 SNP markers
| Chromosome | N˚Markers | Chromosome | N˚Markers | Chromosome | N˚Markers |
|---|---|---|---|---|---|
| Chr 1 | 36,510 | Chr 7 | 15,271 | Chr 13 | 18,743 |
| Chr 2 | 28,503 | Chr 8 | 21,154 | Chr 14 | 24,541 |
| Chr 3 | 25,488 | Chr 9 | 21,216 | Chr 15 | 23,530 |
| Chr 4 | 22,497 | Chr 10 | 19,127 | Chr 16 | 18,031 |
| Chr 5 | 24,234 | Chr 11 | 22,587 | Chr 17 | 18,385 |
| Chr 6 | 23,841 | Chr 12 | 18,705 | Chr 18 | 17,759 |
library(here); library(tidyverse)here() starts at /Users/lbd54/Documents/GitHub/EMBRAPAImputation2022── Attaching packages ─────────────────────────────────────── tidyverse 1.3.1 ──✔ ggplot2 3.3.6 ✔ purrr 0.3.4
✔ tibble 3.1.7 ✔ dplyr 1.0.9
✔ tidyr 1.2.0 ✔ stringr 1.4.0
✔ readr 2.1.2 ✔ forcats 0.5.1── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()GBSVCFFile <- read.table(file = here::here("data", "GBS", "tassel_embrapa_newfastq.bial.recode.vcf"),
comment.char = "", header = T, skip = 31)
GBSMkrs <- GBSVCFFile %>% select(ID, REF, ALT, X.CHROM, POS) %>%
mutate(IDPol = paste0(ID,"_", REF, "/", ALT))
write.table(x = GBSMkrs, file = here::here("output", "DArT2022", "GBSMkrs.txt"),
quote = F, row.names = F)
GBSMkrs <- read.table(file = here::here("output", "DArT2022", "GBSMkrs.txt"), header = T)
DArTMkrs <- read.table(file = here::here("output", "DArT2022", "DArTMkrs.txt"), header = T)
CommonMkrs <- list(DArT = DArTMkrs$IDPol,
GBS = GBSMkrs$IDPol)
CMkrs <- inner_join(DArTMkrs, GBSMkrs, by = c("IDPol"), suffix = c("DArT", "GBS"))
CMkrs2 <- CMkrs %>% dplyr::select(IDDArT, REFDArT, ALTDArT, X.CHROMDArT, POS) %>%
rename(ID = IDDArT,
REF = REFDArT,
ALT = ALTDArT,
Chr = X.CHROMDArT,
Pos = POS)
write.table(CMkrs2, file = here::here("data", "CommonMkrsGBSDArT.txt"), row.names = F,
quote = F, sep = "\t")
write.table(CMkrs2 %>% dplyr::select(Chr, Pos), file = here::here("data", "CommonMkrsGBSDArT"),
row.names = F, col.names = F, quote = F, sep = "\t")
CMkrsIssues <- inner_join(DArTMkrs, GBSMkrs, by = c("ID"),
suffix = c("DArT", "GBS")) %>%
filter(IDPolDArT != IDPolGBS)
write.table(CMkrsIssues %>% dplyr::select(X.CHROMGBS, POS),
file = here::here("data", "CommonMkrsGBSDArTIssues"),
row.names = F, col.names = F, quote = F, sep = "\t")Fig 1. Venn Diagram of the common SNP markers between the GBS and DArT genotyping platforms
Loading required package: grid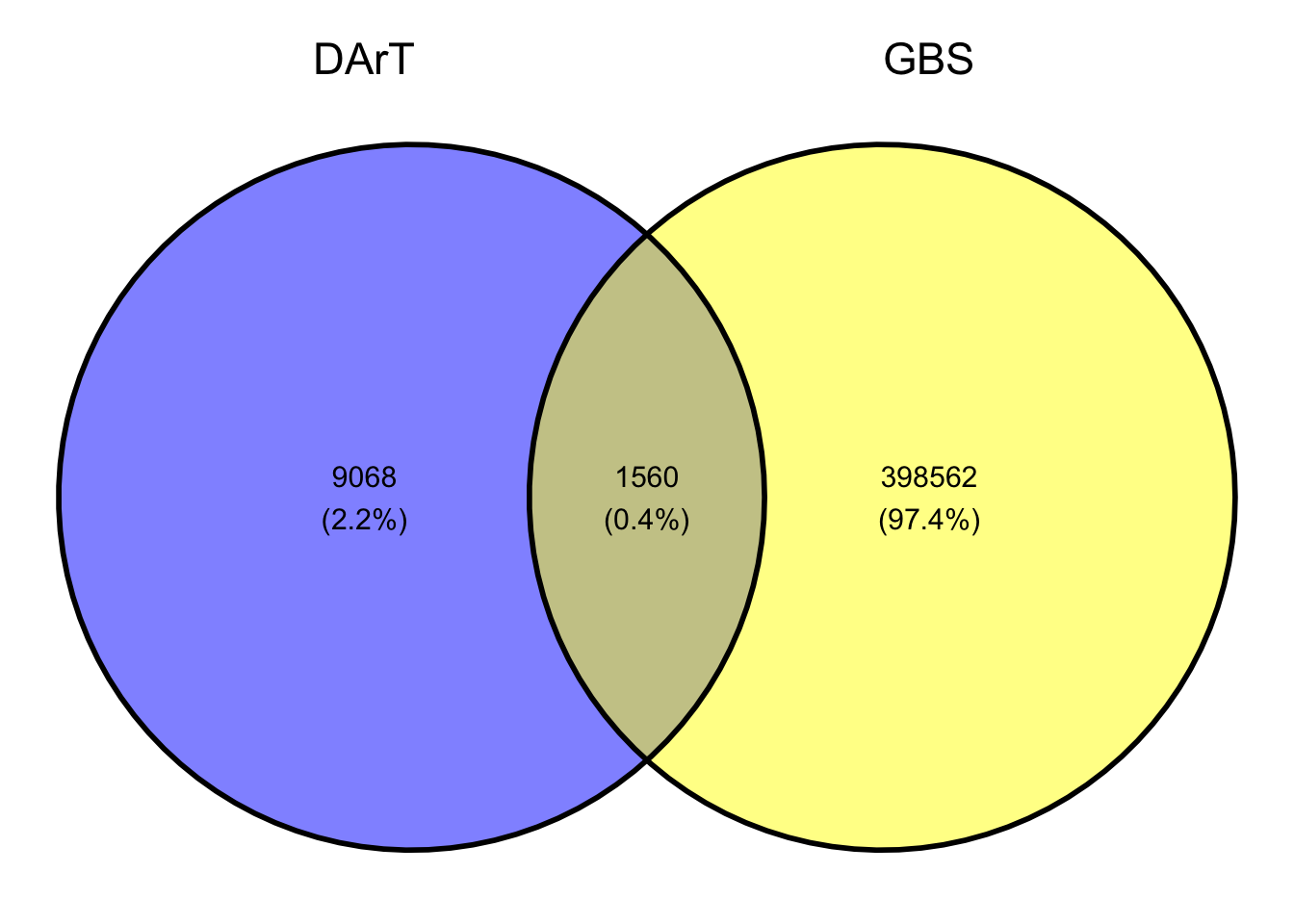
| Chr | N˚SNP only DArT | N˚ DArT + GBS | N˚SNP GBS |
|---|---|---|---|
| 1 | 881 | 183 | 36,327 |
| 2 | 593 | 103 | 28,400 |
| 3 | 581 | 101 | 25,387 |
| 4 | 588 | 94 | 22,403 |
| 5 | 533 | 101 | 24,133 |
| 6 | 552 | 104 | 23,737 |
| 7 | 366 | 62 | 15,209 |
| 8 | 438 | 94 | 21,060 |
| 9 | 431 | 89 | 21,127 |
| 10 | 577 | 87 | 19,040 |
| 11 | 507 | 84 | 22,503 |
| 12 | 381 | 87 | 18,618 |
| 13 | 434 | 74 | 18,669 |
| 14 | 592 | 115 | 24,426 |
| 15 | 435 | 81 | 23,449 |
| 16 | 360 | 77 | 17,954 |
| 17 | 464 | 79 | 18,306 |
| 18 | 408 | 72 | 17,687 |
| Total | 10,628 | 1,560 | 400,122 |
vcftools --gzvcf output/DArT2022/DCas22_6902_DArTseqLD_AllSites_AllChrom_raw.vcf.gz --positions data/CommonMkrsGBSDArT --recode --stdout | bgzip -c -@ 3 > output/DCas22_6902/DArTCommonMkrs.vcf.gz
vcftools --gzvcf data/GBS/tassel_embrapa_newfastq.bial.recode.vcf.gz --positions data/CommonMkrsGBSDArT --recode --stdout | bgzip -c -@ 3 > output/DCas22_6902/GBSCommonMkrs.vcf.gzVerify the Common Clones genotyped
GBSClones <- read.table(file = here::here("data", "GBS", "EM_matched_accessions-GBS.csv"),
sep = ",", header = T)
## Duplicates in GBS genotyping platform
dup <- GBSClones[GBSClones$uniquename_in_db %in%
GBSClones$uniquename_in_db[duplicated(GBSClones$uniquename_in_db)],] %>%
.[order(.[,2]),]
dup1 <- GBSClones[duplicated(GBSClones$uniquename_in_db),] %>% .[order(.[,2]),]
dup2 <- dup[!dup$x %in% dup1$x,] %>%
.[order(.[,2]),] %>% mutate(FILE = "2")
dup3 <- dup1[duplicated(dup1$uniquename_in_db),] %>%
.[order(.[,2]),] %>% mutate(FILE = "3")
dup1 <- dup1[!dup1$x %in% dup3$x,] %>% mutate(FILE = "1")
write.table(x = rbind(dup1, dup2, dup3), quote = F, row.names = F, sep = "\t",
file = here::here("data", "GBS", "AllGBSDuplicates.txt"))
write.table(x = dup1$x, file = here::here("data", "GBS", "GBSDuplicates1.txt"),
sep = "\t", row.names = F, col.names = F, quote = F)
write.table(x = dup2$x, file = here::here("data", "GBS", "GBSDuplicates2.txt"),
sep = "\t", row.names = F, col.names = F, quote = F)
write.table(x = dup3$x, file = here::here("data", "GBS", "GBSDuplicates3.txt"),
sep = "\t", row.names = F, col.names = F, quote = F)
### Marnin had an excellent idea of sums the counts for each allele for the duplicates individuals
## Duplicates in DArT genotyping platform
DArTClones <- read.table(file = here::here("data", "DArTGPInfo2.csv"),
sep = ",", header = T)
Dup <- DArTClones[DArTClones$Observation_unit_name %in%
DArTClones$Observation_unit_name[duplicated(DArTClones$Observation_unit_name)],]
Dup1 <- DArTClones %>% filter(duplicated(DArTClones$Observation_unit_name)) %>%
filter(!duplicated(.[,"Observation_unit_name"])) %>%
.[order(.[,"Observation_unit_name"]),] %>% mutate(FILE = "1")
Dup2 <- Dup %>% filter(!Dup$exported_tissue_sample_name %in%
Dup1$exported_tissue_sample_name) %>%
filter(!duplicated(.[,"Observation_unit_name"])) %>% mutate(FILE = "2")
Dup3 <- Dup %>% filter(!Dup$exported_tissue_sample_name %in%
(rbind(Dup1, Dup2) %>%
.[,"exported_tissue_sample_name"])) %>%
filter(!duplicated(.[,"Observation_unit_name"])) %>% mutate(FILE = "3")
Dup4 <- Dup %>% filter(!Dup$exported_tissue_sample_name %in%
(rbind(Dup1, Dup2, Dup3) %>%
.[,"exported_tissue_sample_name"])) %>%
mutate(FILE = "4")
write.table(x = rbind(Dup1, Dup2, Dup3, Dup4), quote = F, row.names = F, sep = "\t",
file = here::here("data", "AllDArTDuplicates.txt"))
write.table(x = Dup1$DarTMatch, file = here::here("data", "DArTDuplicates1.txt"),
sep = "\t", row.names = F, col.names = F, quote = F)
write.table(x = Dup2$DarTMatch, file = here::here("data", "DArTDuplicates2.txt"),
sep = "\t", row.names = F, col.names = F, quote = F)
write.table(x = Dup3$DarTMatch, file = here::here("data", "DArTDuplicates3.txt"),
sep = "\t", row.names = F, col.names = F, quote = F)
write.table(x = Dup4$DarTMatch, file = here::here("data", "DArTDuplicates4.txt"),
sep = "\t", row.names = F, col.names = F, quote = F)Check the duplicates in each genotyping platform link
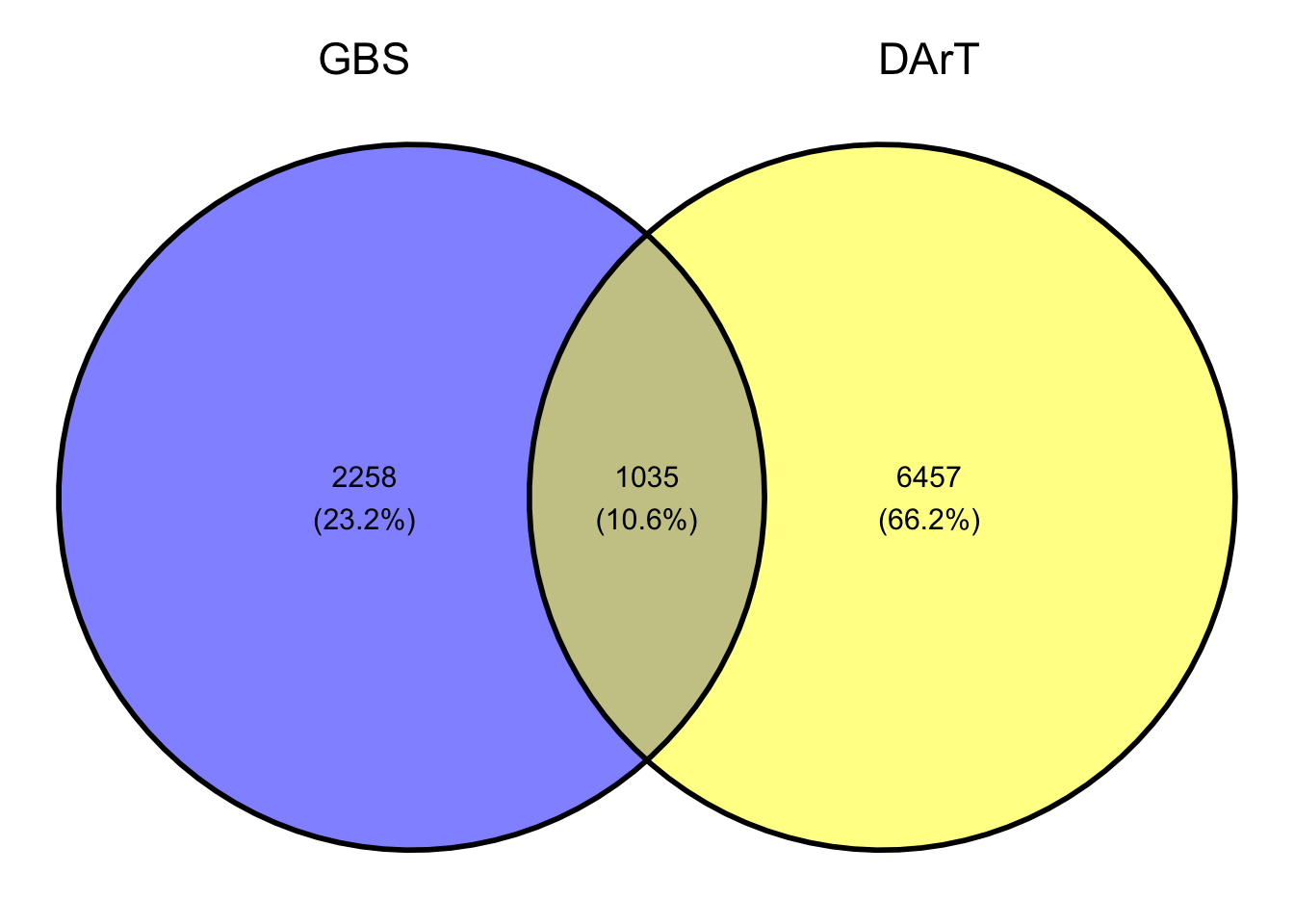
Fig 2. Venn Diagram of the common clones between the GBS and DArT genotyping platforms
CommonClones <- GBSClones$uniquename_in_db[GBSClones$uniquename_in_db %in%
DArTClones$Observation_unit_name] %>%
unique
GBSComClones <- GBSClones[GBSClones$uniquename_in_db %in% CommonClones,] %>%
arrange(uniquename_in_db)
DArTComClones <- DArTClones[DArTClones$Observation_unit_name %in% CommonClones,] %>%
arrange(Observation_unit_name)
GBSComClones1 <- GBSComClones %>% filter(!duplicated(GBSComClones$uniquename_in_db)) %>%
.[order(.[,"uniquename_in_db"]),] %>% mutate(FILE = "G1")
GBSComClones2 <- GBSComClones %>% filter(!GBSComClones$x %in% GBSComClones1$x) %>%
filter(!duplicated(.[,"uniquename_in_db"])) %>% .[order(.[,"uniquename_in_db"]),] %>%
mutate(FILE = "G2")
GBSComClones3 <- GBSComClones %>% filter(!GBSComClones$x %in%
(rbind(GBSComClones1, GBSComClones2) %>%
.[,"x"])) %>%
filter(!duplicated(.[,"uniquename_in_db"])) %>% .[order(.[,"uniquename_in_db"]),] %>%
mutate(FILE = "G3")
write.table(rbind(GBSComClones1, GBSComClones2, GBSComClones3),
file = here::here("output", "DCas22_6902", "AllGBSComClones.txt"),
quote = F, sep = "\t")
write.table(GBSComClones1$x, file = here::here("output", "DCas22_6902", "GBSComClones1"),
row.names = F, col.names = F, quote = F, sep = "\t")
write.table(GBSComClones2$x, file = here::here("output", "DCas22_6902", "GBSComClones2"),
row.names = F, col.names = F, quote = F, sep = "\t")
write.table(GBSComClones3$x, file = here::here("output", "DCas22_6902", "GBSComClones3"),
row.names = F, col.names = F, quote = F, sep = "\t")
DArTComClones1 <- DArTComClones %>% filter(!duplicated(DArTComClones$Observation_unit_name)) %>%
.[order(.[,"Observation_unit_name"]),] %>% mutate(FILE = "D1")
DArTComClones2 <- DArTComClones %>% filter(!DArTComClones$DarTMatch %in% DArTComClones1$DarTMatch) %>%
filter(!duplicated(.[,"Observation_unit_name"])) %>% .[order(.[,"Observation_unit_name"]),] %>%
mutate(FILE = "D2")
DArTComClones3 <- DArTComClones %>% filter(!DArTComClones$DarTMatch %in%
(rbind(DArTComClones1, DArTComClones2) %>%
.[,"DarTMatch"])) %>%
filter(!duplicated(.[,"Observation_unit_name"])) %>% .[order(.[,"Observation_unit_name"]),] %>%
mutate(FILE = "D3")
DArTComClones4 <- DArTComClones %>% filter(!DArTComClones$DarTMatch %in%
(rbind(DArTComClones1, DArTComClones2, DArTComClones3) %>%
.[,"DarTMatch"])) %>%
filter(!duplicated(.[,"Observation_unit_name"])) %>% .[order(.[,"Observation_unit_name"]),] %>%
mutate(FILE = "D4")
write.table(rbind(DArTComClones1, DArTComClones2, DArTComClones3, DArTComClones4),
file = here::here("output", "DCas22_6902", "AllDArTComClones.txt"),
quote = F, sep = "\t")
write.table(DArTComClones1$DarTMatch, file = here::here("output", "DCas22_6902", "DArTComClones1"),
row.names = F, col.names = F, quote = F, sep = "\t")
write.table(DArTComClones2$DarTMatch, file = here::here("output", "DCas22_6902", "DArTComClones2"),
row.names = F, col.names = F, quote = F, sep = "\t")
write.table(DArTComClones3$DarTMatch, file = here::here("output", "DCas22_6902", "DArTComClones3"),
row.names = F, col.names = F, quote = F, sep = "\t")
write.table(DArTComClones4$DarTMatch, file = here::here("output", "DCas22_6902", "DArTComClones4"),
row.names = F, col.names = F, quote = F, sep = "\t")### GBS Selecting files
system(paste0("vcftools --gzvcf output/DCas22_6902/GBSCommonMkrs.vcf.gz --keep output/DCas22_6902/GBSComClones", 1:3, " --recode --stdout > output/DCas22_6902/GBSComClones", 1:3, ".vcf"))
### DArT Selecting files
system(paste0("vcftools --gzvcf output/DCas22_6902/DArTCommonMkrs.vcf.gz --keep output/DCas22_6902/DArTComClones", 1:4, " --recode --stdout > output/DCas22_6902/DArTComClones", 1:4, ".vcf"))### GBS
GBSC1 <- read.table(file = here::here("output", "DCas22_6902", "GBSComClones1.vcf"),
skip = 31, header = T, comment.char = "", check.names = F)
GBSC2 <- read.table(file = here::here("output", "DCas22_6902", "GBSComClones2.vcf"),
skip = 31, header = T, comment.char = "", check.names = F)
GBSC3 <- read.table(file = here::here("output", "DCas22_6902", "GBSComClones3.vcf"),
skip = 31, header = T, comment.char = "", check.names = F)
GBSClones <- read.table(file = here::here("data", "GBS", "EM_matched_accessions-GBS.csv"),
sep = ",", header = T)
colnames(GBSC1)[10:ncol(GBSC1)] <- GBSClones$uniquename_in_db[match(colnames(GBSC1)[10:ncol(GBSC1)], GBSClones$x)]
colnames(GBSC2)[10:ncol(GBSC2)] <- GBSClones$uniquename_in_db[match(colnames(GBSC2)[10:ncol(GBSC2)], GBSClones$x)]
colnames(GBSC3)[10:ncol(GBSC3)] <- GBSClones$uniquename_in_db[match(colnames(GBSC3)[10:ncol(GBSC3)], GBSClones$x)]
header <- c("##fileformat=VCFv4.0",
"##FILTER=<ID=PASS,Description=\"All filters passed\">",
"##Tassel=<ID=GenotypeTable,Version=5,Description=\"Reference allele is not known. The major allele was used as reference allele\">",
"##FORMAT=<ID=GT,Number=1,Type=String,Description=\"Genotype\">",
"##FORMAT=<ID=AD,Number=.,Type=Integer,Description=\"Allelic depths for the reference and alternate alleles in the order listed\">",
"##FORMAT=<ID=DP,Number=1,Type=Integer,Description=\"Read Depth (only filtered reads used for calling)\">",
"##FORMAT=<ID=GQ,Number=1,Type=Float,Description=\"Genotype Quality\">",
"##FORMAT=<ID=PL,Number=.,Type=Float,Description=\"Normalized, Phred-scaled likelihoods for AA,AB,BB genotypes where A=ref and B=alt; not applicable if site is not biallelic\">",
"##INFO=<ID=NS,Number=1,Type=Integer,Description=\"Number of Samples With Data\">",
"##INFO=<ID=DP,Number=1,Type=Integer,Description=\"Total Depth\">",
"##INFO=<ID=AF,Number=.,Type=Float,Description=\"Allele Frequency\">",
"##contig=<ID=1>",
"##contig=<ID=2>",
"##contig=<ID=3>",
"##contig=<ID=4>",
"##contig=<ID=5>",
"##contig=<ID=6>",
"##contig=<ID=7>",
"##contig=<ID=8>",
"##contig=<ID=9>",
"##contig=<ID=10>",
"##contig=<ID=11>",
"##contig=<ID=12>",
"##contig=<ID=13>",
"##contig=<ID=14>",
"##contig=<ID=15>",
"##contig=<ID=16>",
"##contig=<ID=17>",
"##contig=<ID=18>",
"##bcftools_concatVersion=1.14+htslib-1.14",
"##bcftools_concatCommand=concat --output tassel_embrapa_newfastq.bial.recode.vcf")
write_lines(header, file = here::here("output", "DCas22_6902", "GBSComClones1renamed.vcf"),
append = F)
write_lines(header, file = here::here("output", "DCas22_6902", "GBSComClones2renamed.vcf"),
append = F)
write_lines(header, file = here::here("output", "DCas22_6902", "GBSComClones3renamed.vcf"),
append = F)
write.table(x = GBSC1, file = here::here("output", "DCas22_6902", "GBSComClones1renamed.vcf"),
quote = F, row.names = F, append = T, col.names = T, sep = "\t")
write.table(x = GBSC2, file = here::here("output", "DCas22_6902", "GBSComClones2renamed.vcf"),
quote = F, row.names = F, append = T, col.names = T, sep = "\t")
write.table(x = GBSC3, file = here::here("output", "DCas22_6902", "GBSComClones3renamed.vcf"),
quote = F, row.names = F, append = T, col.names = T, sep = "\t")
### DArT
DArTC1 <- read.table(file = here::here("output", "DCas22_6902", "DArTComClones1.vcf"),
header = T, comment.char = "", check.names = F)
DArTC2 <- read.table(file = here::here("output", "DCas22_6902", "DArTComClones2.vcf"),
header = T, comment.char = "", check.names = F)
DArTC3 <- read.table(file = here::here("output", "DCas22_6902", "DArTComClones3.vcf"),
header = T, comment.char = "", check.names = F)
DArTC4 <- read.table(file = here::here("output", "DCas22_6902", "DArTComClones4.vcf"),
header = T, comment.char = "", check.names = F)
DArTClones <- read.table(file = here::here("data", "DArTGPInfo2.csv"),
sep = ",", header = T)
colnames(DArTC1)[10:ncol(DArTC1)] <- DArTClones$Observation_unit_name[match(colnames(DArTC1)[10:ncol(DArTC1)], DArTClones$DarTMatch)]
colnames(DArTC2)[10:ncol(DArTC2)] <- DArTClones$Observation_unit_name[match(colnames(DArTC2)[10:ncol(DArTC2)], DArTClones$DarTMatch)]
colnames(DArTC3)[10:ncol(DArTC3)] <- DArTClones$Observation_unit_name[match(colnames(DArTC3)[10:ncol(DArTC3)], DArTClones$DarTMatch)]
colnames(DArTC4)[10:ncol(DArTC4)] <- DArTClones$Observation_unit_name[match(colnames(DArTC4)[10:ncol(DArTC4)], DArTClones$DarTMatch)]
header <- c("##fileformat=VCFv4.0",
"##FORMAT=<ID=GT,Number=1,Type=String,Description=\"Genotype\">",
"##FORMAT=<ID=AD,Number=.,Type=Integer,Description=\"Allelic depths for the reference and alternate alleles in the order listed\">",
"##FORMAT=<ID=DP,Number=1,Type=Integer,Description=\"Read Depth (only filtered reads used for calling)\">",
"##FORMAT=<ID=PL,Number=3,Type=Float,Description=\"Normalized, Phred-scaled likelihoods for AA,AB,BB genotypes where A=ref and B=alt; not applicable if site is not biallelic\">")
write_lines(header, file = here::here("output", "DCas22_6902", "DArTComClones1renamed.vcf"),
append = F)
write_lines(header, file = here::here("output", "DCas22_6902", "DArTComClones2renamed.vcf"),
append = F)
write_lines(header, file = here::here("output", "DCas22_6902", "DArTComClones3renamed.vcf"),
append = F)
write_lines(header, file = here::here("output", "DCas22_6902", "DArTComClones4renamed.vcf"),
append = F)
write.table(x = DArTC1, file = here::here("output", "DCas22_6902", "DArTComClones1renamed.vcf"),
quote = F, row.names = F, append = T, col.names = T, sep = "\t")
write.table(x = DArTC2, file = here::here("output", "DCas22_6902", "DArTComClones2renamed.vcf"),
quote = F, row.names = F, append = T, col.names = T, sep = "\t")
write.table(x = DArTC3, file = here::here("output", "DCas22_6902", "DArTComClones3renamed.vcf"),
quote = F, row.names = F, append = T, col.names = T, sep = "\t")
write.table(x = DArTC4, file = here::here("output", "DCas22_6902", "DArTComClones4renamed.vcf"),
quote = F, row.names = F, append = T, col.names = T, sep = "\t")source(paste0("bgzip -c -@ 6 output/DCas22_6902/GBSComClones", 1:3, "renamed.vcf > output/DCas22_6902/GBSComClones", 1:3, "renamed.vcf.gz"))
source(paste0("bgzip -c -@ 6 output/DCas22_6902/DArTComClones", 1:4, "renamed.vcf > output/DCas22_6902/DArTComClones", 1:4, "renamed.vcf.gz"))Comparing the Duplicates
vcftools --gzvcf output/DCas22_6902/GBSComClones1renamed.vcf.gz --gzdiff \
output/DCas22_6902/DArTComClones1renamed.vcf.gz --diff-indv-discordance --out output/DCas22_6902/GBSDArT1_1
vcftools --gzvcf output/DCas22_6902/GBSComClones1renamed.vcf.gz --gzdiff \
output/DCas22_6902/DArTComClones2renamed.vcf.gz --diff-indv-discordance --out output/DCas22_6902/GBSDArT1_2
vcftools --gzvcf output/DCas22_6902/GBSComClones1renamed.vcf.gz --gzdiff \
output/DCas22_6902/DArTComClones3renamed.vcf.gz --diff-indv-discordance --out output/DCas22_6902/GBSDArT1_3
vcftools --gzvcf output/DCas22_6902/GBSComClones1renamed.vcf.gz --gzdiff \
output/DCas22_6902/DArTComClones4renamed.vcf.gz --diff-indv-discordance --out output/DCas22_6902/GBSDArT1_4
vcftools --gzvcf output/DCas22_6902/GBSComClones2renamed.vcf.gz --gzdiff \
output/DCas22_6902/DArTComClones1renamed.vcf.gz --diff-indv-discordance --out output/DCas22_6902/GBSDArT2_1
vcftools --gzvcf output/DCas22_6902/GBSComClones2renamed.vcf.gz --gzdiff \
output/DCas22_6902/DArTComClones2renamed.vcf.gz --diff-indv-discordance --out output/DCas22_6902/GBSDArT2_2
vcftools --gzvcf output/DCas22_6902/GBSComClones2renamed.vcf.gz --gzdiff \
output/DCas22_6902/DArTComClones3renamed.vcf.gz --diff-indv-discordance --out output/DCas22_6902/GBSDArT2_3
vcftools --gzvcf output/DCas22_6902/GBSComClones2renamed.vcf.gz --gzdiff \
output/DCas22_6902/DArTComClones4renamed.vcf.gz --diff-indv-discordance --out output/DCas22_6902/GBSDArT2_4
vcftools --gzvcf output/DCas22_6902/GBSComClones3renamed.vcf.gz --gzdiff \
output/DCas22_6902/DArTComClones1renamed.vcf.gz --diff-indv-discordance --out output/DCas22_6902/GBSDArT3_1
vcftools --gzvcf output/DCas22_6902/GBSComClones3renamed.vcf.gz --gzdiff \
output/DCas22_6902/DArTComClones2renamed.vcf.gz --diff-indv-discordance --out output/DCas22_6902/GBSDArT3_2
vcftools --gzvcf output/DCas22_6902/GBSComClones3renamed.vcf.gz --gzdiff \
output/DCas22_6902/DArTComClones3renamed.vcf.gz --diff-indv-discordance --out output/DCas22_6902/GBSDArT3_3
vcftools --gzvcf output/DCas22_6902/GBSComClones3renamed.vcf.gz --gzdiff \
output/DCas22_6902/DArTComClones4renamed.vcf.gz --diff-indv-discordance --out output/DCas22_6902/GBSDArT3_4Joint the results
library(tidyverse); library(magrittr)
Attaching package: 'magrittr'The following object is masked from 'package:purrr':
set_namesThe following object is masked from 'package:tidyr':
extractGBSDArT3_4 <- read.table(file = here::here("output", "DCas22_6902", "GBSDArT1_1.diff.indv"),
sep = "\t", header = T, check.names = F) %>%
mutate(COMP1 = "G1",
COMP2 = "D1") %>%
rbind(read.table(file = here::here("output", "DCas22_6902", "GBSDArT1_2.diff.indv"),
sep = "\t", header = T, check.names = F) %>%
mutate(COMP1 = "G1",
COMP2 = "D2")) %>%
rbind(read.table(file = here::here("output", "DCas22_6902", "GBSDArT1_3.diff.indv"),
sep = "\t", header = T, check.names = F) %>%
mutate(COMP1 = "G1",
COMP2 = "D3")) %>%
rbind(read.table(file = here::here("output", "DCas22_6902", "GBSDArT1_4.diff.indv"),
sep = "\t", header = T, check.names = F) %>%
mutate(COMP1 = "G1",
COMP2 = "D4")) %>%
rbind(read.table(file = here::here("output", "DCas22_6902", "GBSDArT2_1.diff.indv"),
sep = "\t", header = T, check.names = F) %>%
mutate(COMP1 = "G2",
COMP2 = "D1")) %>%
rbind(read.table(file = here::here("output", "DCas22_6902", "GBSDArT2_2.diff.indv"),
sep = "\t", header = T, check.names = F) %>%
mutate(COMP1 = "G2",
COMP2 = "D2")) %>%
rbind(read.table(file = here::here("output", "DCas22_6902", "GBSDArT2_3.diff.indv"),
sep = "\t", header = T, check.names = F) %>%
mutate(COMP1 = "G2",
COMP2 = "D3")) %>%
rbind(read.table(file = here::here("output", "DCas22_6902", "GBSDArT2_4.diff.indv"),
sep = "\t", header = T, check.names = F) %>%
mutate(COMP1 = "G2",
COMP2 = "D4")) %>%
rbind(read.table(file = here::here("output", "DCas22_6902", "GBSDArT3_1.diff.indv"),
sep = "\t", header = T, check.names = F) %>%
mutate(COMP1 = "G3",
COMP2 = "D1")) %>%
rbind(read.table(file = here::here("output", "DCas22_6902", "GBSDArT3_2.diff.indv"),
sep = "\t", header = T, check.names = F) %>%
mutate(COMP1 = "G3",
COMP2 = "D2")) %>%
rbind(read.table(file = here::here("output", "DCas22_6902", "GBSDArT3_3.diff.indv"),
sep = "\t", header = T, check.names = F) %>%
mutate(COMP1 = "G3",
COMP2 = "D3")) %>%
rbind(read.table(file = here::here("output", "DCas22_6902", "GBSDArT3_4.diff.indv"),
sep = "\t", header = T, check.names = F) %>%
mutate(COMP1 = "G3",
COMP2 = "D4")) %>%
filter(!is.na(DISCORDANCE)) %>%
mutate(N_NA = 1560 - N_COMMON_CALLED) %>%
select(INDV, COMP1, COMP2, N_NA, N_COMMON_CALLED, N_DISCORD, DISCORDANCE) %>% arrange(INDV, COMP1, COMP2)
### Check the names in each vcf file
AllGBSComCl <- read.table(file = here::here("output", "DCas22_6902", "AllGBSComClones.txt"),
header = T, sep = "\t")
AllDArTComCl <- read.table(file = here::here("output", "DCas22_6902", "AllDArTComClones.txt"),
header = T, sep = "\t")
GBSDArT3_4 %<>% left_join(AllGBSComCl, by = c("INDV" = "uniquename_in_db", "COMP1" = "FILE")) %>%
rename(INDV1 = x) %>% left_join(AllDArTComCl, by = c("INDV" = "Observation_unit_name", "COMP2" = "FILE")) %>%
rename(INDV2 = DarTMatch) %>% select(INDV, INDV1, INDV2, N_NA, N_COMMON_CALLED, N_DISCORD, DISCORDANCE)
write.table(GBSDArT3_4, file = here::here("output", "DCas22_6902", "GBSDArTAllDiscordanceIndex.csv"),
quote = F, sep = ",", dec = ".", row.names = F)
DupGBSDArT3_4Cl <- GBSDArT3_4 %>% select(INDV) %>%
.[duplicated(.[,1]),] %>% unique
UnGBSDArT3_4 <- GBSDArT3_4 %>%
.[!.[,"INDV"] %in% DupGBSDArT3_4Cl,] %>% arrange(INDV)
DupGBSDArT3_4 <- GBSDArT3_4 %>% arrange(INDV, DISCORDANCE) %>%
group_by(INDV) %>% slice_min(DISCORDANCE, n = 1) %>% filter(INDV2 != "BGM.0779.1")
DupGBSDArT3_4[duplicated(DupGBSDArT3_4$INDV),]# A tibble: 0 × 7
# Groups: INDV [0]
# … with 7 variables: INDV <chr>, INDV1 <chr>, INDV2 <chr>, N_NA <dbl>,
# N_COMMON_CALLED <int>, N_DISCORD <int>, DISCORDANCE <dbl>DupGBSDArT3_4[DupGBSDArT3_4$INDV %in% "BGM-0779",]# A tibble: 1 × 7
# Groups: INDV [1]
INDV INDV1 INDV2 N_NA N_COMMON_CALLED N_DISCORD DISCORDANCE
<chr> <chr> <chr> <dbl> <int> <int> <dbl>
1 BGM-0779 CNPMF779:250370902 BGM.0… 845 715 220 0.308length(unique(DupGBSDArT3_4$INDV2))[1] 1036write.table(DupGBSDArT3_4, file = here::here("output", "DCas22_6902", "GBSDArTCommonClonesSelected.csv"),
quote = F, sep = ",", dec = ".", row.names = F)
write.table(DupGBSDArT3_4$INDV1, file = here::here("output", "DCas22_6902", "GBSCommonClonesSelected.txt"),
quote = F, sep = "\t", row.names = F, col.names = F)
write.table(DupGBSDArT3_4$INDV2, file = here::here("output", "DCas22_6902", "DArTCommonClonesSelected.txt"),
quote = F, sep = "\t", row.names = F, col.names = F)Table 2. Discordance index from vcftools software for all the duplicates in DArT genotyping platform.
Preparing VCF Files to check Clones genotyped by both Genotyping Platforms
VCFFiles <- c("GBSCommonMkrs.vcf.gz",
"DArTCommonMkrs.vcf.gz")
IndFiles <- c("GBSCommonClonesSelected.txt",
"DArTCommonClonesSelected.txt")
OutFiles <- c("GBSCommonMkrsClones.vcf.gz",
"DArTCommonMkrsClones.vcf.gz")
system(paste0("vcftools --gzvcf output/DCas22_6902/", VCFFiles, " --keep output/DCas22_6902/",
IndFiles, " --recode --stdout | bgzip -c -@ 4 > output/DCas22_6902/",
OutFiles))
system("gzcat output/DCas22_6902/GBSCommonMkrsClones.vcf.gz | head -n31 > output/DCas22_6902/HeaderGBS.txt")
system("bgzip -d output/DCas22_6902/DArTCommonMkrsClones.vcf.gz")
header <- c("##fileformat=VCFv4.0",
"##FORMAT=<ID=GT,Number=1,Type=String,Description=\"Genotype\">",
"##FORMAT=<ID=AD,Number=.,Type=Integer,Description=\"Allelic depths for the reference and alternate alleles in the order listed\">",
"##FORMAT=<ID=DP,Number=1,Type=Integer,Description=\"Read Depth (only filtered reads used for calling)\">",
"##FORMAT=<ID=PL,Number=3,Type=Float,Description=\"Normalized, Phred-scaled likelihoods for AA,AB,BB genotypes where A=ref and B=alt; not applicable if site is not biallelic\">")
DArTVCFFileSel <- read.table(file = here::here("output", "DCas22_6902", "DArTCommonMkrsClones.vcf"),
sep = "\t", header = T, comment.char = "", check.names = F)
write_lines(header, file = here::here("output", "DCas22_6902", "DArTCommonMkrsClones2.vcf"),
append = F)
write.table(x = DArTVCFFileSel, file = here::here("output", "DCas22_6902", "DArTCommonMkrsClones2.vcf"),
quote = F, row.names = F, append = T, col.names = T, sep = "\t")
system(paste0("bgzip -c -@ 4 output/DCas22_6902/DArTCommonMkrsClones2.vcf > ",
"output/DCas22_6902/DArTCommonMkrsClones2.vcf.gz"))system("tabix -f -p vcf output/DCas22_6902/GBSCommonMkrsClones.vcf.gz")
system("tabix -f -p vcf output/DCas22_6902/DArTCommonMkrsClones2.vcf.gz")Merging DArT-GBS VCF files
system(paste0("bcftools merge",
" --output output/DCas22_6902/AllSamplesGBSDArTCommonSites.vcf.gz",
" --merge snps --output-type z --threads 4",
" output/DCas22_6902/GBSCommonMkrsClones.vcf.gz",
" output/DCas22_6902/DArTCommonMkrsClones2.vcf.gz"))Pre-analysis Filtering
system(paste0("vcftools --gzvcf output/DCas22_6902/AllSamplesGBSDArTCommonSites.vcf.gz ",
"--min-alleles 2 --max-alleles 2 --minDP 4, --maxDP 50 ",
"--recode --stdout | bgzip -c -@ 4 > ",
"output/DCas22_6902/AllSamplesGBSDArTCommonSitesFiltered.vcf.gz"))Make binary plink (bed/bim/fam)
system(paste0("bgzip -d -@ 4 output/DCas22_6902/AllSamplesGBSDArTCommonSitesFiltered.vcf.gz > ",
"output/DCas22_6902/AllSamplesGBSDArTCommonSitesFiltered.vcf"))
system(paste0("code/plink/plink",
" --vcf output/DCas22_6902/AllSamplesGBSDArTCommonSitesFiltered.vcf ",
"--make-bed --const-fid ",
"--out output/DCas22_6902/AllSamplesGBSDArTCommonSitesFiltered.vcf")) Plink IBD
system(paste0("code/plink/plink --bfile output/DCas22_6902/AllSamplesGBSDArTCommonSitesFiltered.vcf ",
"--genome --out output/DCas22_6902/AllSamplesGBSDArTCommonSitesFiltered.vcf;"))Matching results DArT-GBS
library(data.table)
Attaching package: 'data.table'The following objects are masked from 'package:dplyr':
between, first, lastThe following object is masked from 'package:purrr':
transposegenome<-fread("output/DCas22_6902/AllSamplesGBSDArTCommonSitesFiltered.vcf.genome",
stringsAsFactors = F, header = T, check.names = F) %>%
as_tibble
dataset_samples<-read.table(file = here::here("output", "DCas22_6902", "GBSDArTCommonClonesSelected.csv"),
header = T, stringsAsFactors = F, check.names = F, sep = ",")
genome2 <- genome %>% filter(IID1 %in% dataset_samples$INDV1, IID2 %in% dataset_samples$INDV2) %>%
inner_join(dataset_samples, by = c("IID1" = "INDV1", "IID2" = "INDV2")) %>%
select(INDV, everything())
write.table(genome2, file = here::here("output", "DCas22_6902", "IDBPlinkCommonClones.csv"),
sep = ",", row.names = F, quote = F)IDBMatches <- genome2 %>% filter(DST >= 0.9 | PI_HAT >= 0.65) %>%
select(IID1, IID2, DST, PI_HAT) %>% rename(NamesGBS = IID1,
NamesDArT = IID2) %>%
filter(!is.na(DST))
write.table(IDBMatches, file = here::here("output", "DCas22_6902", "CommonClonesVerifiedIBD.csv"),
sep = ",", quote = F, row.names = F)
write.table(IDBMatches$NamesGBS, file = here::here("output", "DCas22_6902", "GBSClonesVerifiedIDB.txt"),
sep = "\t", quote = F, row.names = F, col.names = F)
write.table(IDBMatches$NamesDArT, file = here::here("output", "DCas22_6902", "DArTClonesVerifiedIDB.txt"),
sep = "\t", quote = F, row.names = F, col.names = F)Preparing the VCF files for the imputation by Beagle 4.1
GBSMkrs <- read.table(file = here::here("output", "DArT2022", "GBSMkrs.txt"),
header = T)
DArTMkrs <- read.table(file = here::here("output", "DArT2022", "DArTMkrs.txt"), header = T)
gbs_unique_sites <- GBSMkrs %>% # GBS sites
rename(CHROM = X.CHROM) %>%
anti_join(DArTMkrs, by = c("ID")) %>% # that are unique in GBS
select(CHROM, POS) %>% arrange(CHROM, POS); nrow(gbs_unique_sites) # 398,435 Mkrs GBS only
write.table(gbs_unique_sites,
file = here::here("output", "DCas22_6902", "gbs_unique_sites.txt"),
row.names = F, col.names = F, quote = F)Prepare the Reference Population VCF File
Prepare a VCF file of the GBS with the common clones with DArT
library(tidyverse); library(magrittr); require(furrr)
system(paste0("vcftools --gzvcf data/GBS/tassel_embrapa_newfastq.bial.recode.vcf.gz ",
"--keep output/DCas22_6902/GBSClonesVerifiedIDB.txt ",
"--positions output/DCas22_6902/gbs_unique_sites.txt ",
"--recode ",
"--stdout | bgzip -c -@ 7 > ",
"output/DCas22_6902/",
"GBSsamples_UniqueSites_SelUnfiltered.vcf.gz"))Prepare a VCF file of the DArT with the common clones with GBS
system(paste0("vcftools --gzvcf output/DCas22_6902/",
"DCas22_6902_DArTseqLD_AllSites_AllChrom_raw.vcf.gz ",
"--keep output/DCas22_6902/",
"DArTClonesVerifiedIDB.txt ",
" --recode ",
"--stdout | bgzip -c -@ 7 > ",
"output/DCas22_6902/",
"DArTsamples_UniqueSites_SelUnfiltered.vcf.gz"))Now we need to rename the clones of the GBS VCF file with the same name in DArT, so we can proceed joint these two files.
system(paste0("tabix -f -p vcf output/DCas22_6902/",
"GBSsamples_UniqueSites_SelUnfiltered.vcf.gz"))
system(paste0("tabix -f -p vcf output/DCas22_6902/",
"DArTsamples_UniqueSites_SelUnfiltered.vcf.gz"))GBS
system(paste0("bcftools query --list-samples output/DCas22_6902/",
"GBSsamples_UniqueSites_SelUnfiltered.vcf.gz ",
"> output/DCas22_6902/",
"GBSSelsampleNamesInOrder_GBSUniqueSites_unfiltered.txt"))DArT
system(paste0("bcftools query --list-samples output/DCas22_6902/",
"DArTsamples_UniqueSites_SelUnfiltered.vcf.gz ",
"> output/DCas22_6902/",
"DArTSelsampleNamesInOrder_DArTUniqueSites_unfiltered.txt"))Let’s see what is the match position of the GBS with DArT clones
library(tidyverse); library(magrittr)
GBSSelClones <- read.table(file = here::here("output", "DCas22_6902",
"GBSSelsampleNamesInOrder_GBSUniqueSites_unfiltered.txt"),
header = F, sep = "\t")
DArTSelClones <- read.table(file = here::here("output", "DCas22_6902",
"DArTSelsampleNamesInOrder_DArTUniqueSites_unfiltered.txt"),
header = F, sep = "\t")
GBSDArTCheckNames <- read.table(file = here::here("output", "DCas22_6902",
"CommonClonesVerifiedIBD.csv"),
sep = ",", header = T)
GBSDArTCheckNames %<>% .[match(DArTSelClones$V1, .$NamesDArT),]
NewGBSSelClones <- GBSDArTCheckNames %>%
.[match(GBSSelClones$V1, .$NamesGBS), "NamesDArT"]
write.table(NewGBSSelClones, file = here::here("output", "DCas22_6902",
"NewGBSClonesNames.txt"),
sep = "\t", quote = F, col.names = F, row.names = F)
write.table(GBSDArTCheckNames$NamesDArT,
file = here::here("output", "DCas22_6902", "GBSClonesPos.txt"),
sep = "\t", quote = F, col.names = F, row.names = F)Change the names and the position of the Clones to the same as DArT
system(paste0("bcftools reheader --samples output/DCas22_6902/",
"NewGBSClonesNames.txt ",
"--output output/DCas22_6902/",
"GBSsamples_UniqueSites_SelUnfilteredRenamed.vcf.gz ",
"output/DCas22_6902/",
"GBSsamples_UniqueSites_SelUnfiltered.vcf.gz"))
system(paste0("bcftools view --samples-file ",
"output/DCas22_6902/",
"GBSClonesPos.txt ",
"--output-type z --output-file ",
"output/DCas22_6902/",
"GBSsamples_UniqueSites_SelUnfilteredRenamed2.vcf.gz ",
"output/DCas22_6902/",
"GBSsamples_UniqueSites_SelUnfilteredRenamed.vcf.gz"))
system(paste0("vcftools --gzvcf output/DCas22_6902/",
"GBSsamples_UniqueSites_SelUnfilteredRenamed2.vcf.gz ",
"--exclude-positions output/DCas22_6902/",
"RmMKrsGBSPreImputedFilter.txt",
" --recode ",
"--stdout | bgzip -c -@ 7 > ",
"output/DCas22_6902/",
"GBSsamples_UniqueSites_SelUnfilteredRenamed2.vcf.gz"))Index GBS VCF File before concat with DArT VCF
system(paste0("tabix -f -p vcf output/DCas22_6902/",
"GBSsamples_UniqueSites_SelUnfilteredRenamed2.vcf.gz"))Concat DArT and GBS Mkrs from the common clones
system(paste0("bcftools concat --allow-overlaps ",
"--output output/DCas22_6902/",
"GBSandDArTsitesCommonClones_unfiltered.vcf.gz ",
"--output-type z --threads 7 ",
"output/DCas22_6902/",
"GBSsamples_UniqueSites_SelUnfilteredRenamed2.vcf.gz ",
"output/DCas22_6902/",
"DArTsamples_UniqueSites_SelUnfiltered.vcf.gz"))Reference Population Filters
system(paste0("vcftools --gzvcf output/DCas22_6902/",
"GBSandDArTsitesCommonClones_unfiltered.vcf.gz ",
"--min-alleles 2 --max-alleles 2 --minDP 5 --maxDP 50 ",
"--recode --stdout | bgzip -c -@ 7 > ",
"output/DCas22_6902/",
"GBSandDArTsitesCommonClones_ready2filter.vcf.gz"))
# Estimating Call rate
system(paste0("vcftools --gzvcf ",
"output/DCas22_6902/",
"GBSandDArTsitesCommonClones_ready2filter.vcf.gz ",
"--missing-site ",
"--out output/DCas22_6902/",
"GBSandDArTsitesCommonClones_ready2filter"))
# Estimating mean filtered depth per site
system(paste0("vcftools --gzvcf ",
"output/DCas22_6902/",
"GBSandDArTsitesCommonClones_ready2filter.vcf.gz ",
"--site-mean-depth ",
"--out output/DCas22_6902/",
"GBSandDArTsitesCommonClones_ready2filter"))
# Estimating Hardy-Weinberg equilibrium
system(paste0("vcftools --gzvcf ",
"output/DCas22_6902/",
"GBSandDArTsitesCommonClones_ready2filter.vcf.gz ",
"--hardy ",
"--out output/DCas22_6902/",
"GBSandDArTsitesCommonClones_ready2filter"))stats2filterOn <- read.table(file = here::here("output", "DCas22_6902",
"GBSandDArTsitesCommonClones_ready2filter.lmiss"),
stringsAsFactors = F, header = T) %>%
inner_join(read.table(file = here::here("output", "DCas22_6902",
"GBSandDArTsitesCommonClones_ready2filter.ldepth.mean"),
stringsAsFactors = F, header = T), by = c("CHR" = "CHROM", "POS")) %>%
inner_join(read.table(file = here::here("output", "DCas22_6902",
"GBSandDArTsitesCommonClones_ready2filter.hwe"),
stringsAsFactors = F, header = T), by = c("CHR", "POS"))
PreImputeFilterMkrs <- stats2filterOn %>% filter(F_MISS <= 0.4,
MEAN_DEPTH <= 120,
-log10(P_HWE) < 20) %>%
mutate(ID = paste0(CHR, "_", POS))
NDupMkrs <- PreImputeFilterMkrs[duplicated(PreImputeFilterMkrs$ID),] %>%
group_by(ID) %>% slice(1) %>% ungroup
write.table(NDupMkrs %>% select(CHR, POS),
file = here::here("output", "DCas22_6902", "RmMKrsGBSPreImputedFilter.txt"),
row.names = F, col.names = F, quote = F)
write.table(PreImputeFilterMkrs %>% select(CHR, POS),
file = here::here("output", "DCas22_6902", "MKrsPreImputedFilter.txt"),
row.names = F, col.names = F, quote = F)
system(paste0("vcftools --gzvcf output/DCas22_6902/",
"GBSandDArTsitesCommonClones_ready2filter.vcf.gz ",
"--positions ",
" output/DCas22_6902/",
"MkrsPreImputedFilter.txt ",
"--chr ", 1:18,
" --recode --stdout | ",
"bgzip -c -@ 7 > ",
"output/RefPop/chr", 1:18,
"_GBSandDArTsitesCommonClones_ready2Impute.vcf.gz"))With the filters we got 98,066 SNP Mkrs for the Reference population imputation
runBeagle5Luc <- function(targetVCF, mapFile, outName, nthreads, maxmem = "500g",
impute = TRUE, ne = 1e+05, samplesToExclude = NULL){
system(paste0("java -Xms2g -Xmx", maxmem, " -jar /programs/beagle/beagle.jar ",
"gt=", targetVCF, " ", "map=", mapFile, " ",
"out=", outName, " ", "nthreads=", nthreads,
" impute=", impute, " ne=", ne,
ifelse(!is.null(samplesToExclude),
paste0(" excludesamples=", samplesToExclude), "")))}
targetVCFpath<-here::here("data", "RefPop") # location of the targetVCF
mapPath<-here::here("data", "CassavaGeneticMapV6updated/")
outPath<-here::here("output/")
outSuffix<-"DCas22_6902"
library(tidyverse); library(magrittr);
purrr::map(1:18,
~runBeagle5Luc(targetVCF=paste0(targetVCFpath,"/chr",.,
"_GBSandDArTsitesCommonClones_ready2Impute.vcf.gz"),
mapFile=paste0(mapPath,"chr",.,
"_cassava_cM_pred.v6_91019.map"),
outName=paste0(outPath,"chr",.,
"_GBSandDArTsitesCommonClones_RefPopImputed"),
nthreads=110))Prepare the Population of interest to be imputated
system(paste0("vcftools --gzvcf output/DCas22_6902/",
"GBSandDArTsitesCommonClones_ready2filter.vcf.gz ",
"--positions ",
" output/DCas22_6902/",
"MkrsPreImputedFilter.txt ",
"--chr ", 1:18,
" --recode --stdout | ",
"bgzip -c -@ 7 > ",
"output/RefPop/chr", 1:18,
"_GBSandDArTsitesCommonClones_ready2Impute.vcf.gz"))GBS clones
Prepare a VCF file of the GBS with the common clones with DArT
system(paste0("vcftools --gzvcf data/GBS/tassel_embrapa_newfastq.bial.recode.vcf.gz ",
"--remove output/DCas22_6902/GBSClonesVerifiedIDB.txt ",
"--exclude-positions data/CommonMkrsGBSDArTIssues ",
"--recode ",
"--stdout | bgzip -c -@ 7 > ",
"output/DCas22_6902/",
"GBSonlysamples_UniqueSites_SelUnfiltered.vcf.gz"))DArT clones
Prepare the VCF File of the DArT genotyping clones not present in GBS, with the SNP markers common with the Reference Population.
purrr::map(1:18, ~system(paste0("vcftools --gzvcf output/DCas22_6902/",
"DCas22_6902_DArTseqLD_AllSites_AllChrom_raw.vcf.gz ",
"--remove output/DCas22_6902/",
"DArTClonesVerifiedIDB.txt ",
"--chr ", 1:18,
" --recode ",
"--stdout | bgzip -c -@ 7 > ",
"output/StudyPop/chr",
1:18, "_DArTonlysamples_UniqueSites_Unfiltered.vcf.gz")))Imputation of the Study Population
targetVCFpath <- here::here("data", "StudyPop/") # location of the targetVCF
RefPath <- here::here("output/")
mapPath <- here::here("data", "CassavaGeneticMapV6updated/")
outPath <- here::here("output/")
purrr::map(1:18,
~genomicMateSelectR::runBeagle5(targetVCF=paste0(targetVCFpath,"chr",.,
"_DArTonlysamples_UniqueSites_Unfiltered.vcf.gz"),
refVCF=paste0(RefPath,"chr",.,
"_GBSandDArTsitesCommonClones_RefPopImputed.vcf.gz"),
mapFile=paste0(mapPath,"chr",.,
"_cassava_cM_pred.v6_91019.map"),
outName=paste0(outPath,"chr",.,
"_DCas22_6902_StudyPopimputed"),
nthreads=110))
## Chromosome 4 had issues during the imputation, which required to increase the size of the window in Centimorgans to 731 to finish the imputation. These procedure is okay the window size is an argument in Beagle 5 to reduce the maximum RAM memory demanded for the imputation.
genomicMateSelectR::runBeagle5(targetVCF=paste0(targetVCFpath,"chr4",
"_DArTonlysamples_UniqueSites_Unfiltered.vcf.gz"),
refVCF=paste0(RefPath,"chr4",
"_GBSandDArTsitesCommonClones_RefPopImputed.vcf.gz"),
mapFile=paste0(mapPath,"chr4",
"_cassava_cM_pred.v6_91019.map"),
outName=paste0(outPath,"chr4",
"_DCas22_6902_StudyPopimputed"),
nthreads=110, window = 731)Concatting the Chromosomes VCFs from the Reference and the Study Populations
GBS
## Indexing files
purrr::map(1:18,
~system(paste0("tabix -f -p vcf output/chr",.,
"_GBSandDArTsitesCommonClones_RefPopImputed.vcf.gz")))
system(paste0("bcftools concat ",
"--output ",
"output/AllChrGBSandDArTsitesCommonClones_RefPopImputed.vcf.gz ",
"--output-type z --threads 18 ",
paste0("output/chr",1:18,
"_GBSandDArTsitesCommonClones_RefPopImputed.vcf.gz",
collapse = " ")))
system(paste0("tabix -f -p vcf output/",
"AllChrGBSandDArTsitesCommonClones_RefPopImputed.vcf.gz"))DArT
purrr::map(1:18,
~system(paste0("tabix -f -p vcf output/chr",.,
"_DCas22_6902_StudyPopimputed.vcf.gz")))
system(paste0("bcftools concat ",
"--output ",
"output/AllChrDCas22_6902_StudyPopimputed.vcf.gz ",
"--output-type z --threads 18 ",
paste0("output/chr",1.:18,
"_DCas22_6902_StudyPopimputed.vcf.gz",
collapse = " ")))
system(paste0("tabix -f -p vcf output/",
"AllChrDCas22_6902_StudyPopimputed.vcf.gz"))system(paste0("bcftools merge",
" --output output/",
"DCas22_6902RefPopImputed.vcf.gz",
" --merge snps --output-type z --threads 7",
" output/AllChrGBSandDArTsitesCommonClones_RefPopImputed.vcf.gz",
" output/AllChrDCas22_6902_StudyPopimputed.vcf.gz"))Post Imputation Filter
inPath<-here::here("output/")
outPath<-here::here("output/")
ncores <- 7
nclones <- 7827
require(furrr); options(mc.cores=ncores); plan(multiprocess)
genomicMateSelectR::postImputeFilter(inPath=inPath,
inName=paste0("DCas22_6902RefPopImputed"),
outPath=outPath,
outName=paste0("DCas22_6902_RefandStudyPopimputedAndFiltered"),
DR2thresh = 0, MAFthresh = (1/nclones)*2)Convert VCF to Dosage matrix
genomicMateSelectR::convertVCFtoDosage(pathIn = "output/", pathOut = "output/",
vcfName = "DCas22_6902_RefandStudyPopimputedAndFiltered")
snps <- read.table(file = here::here("output", "DCas22_6902_RefandStudyPopimputedAndFiltered.raw"),
stringsAsFactor=F, header = T) %>%
dplyr::select(-FID,-PAT,-MAT,-SEX,-PHENOTYPE) %>%
column_to_rownames(var = "IID") %>%
as.matrix()
saveRDS(snps,file = here::here("output", "DCas22_GBSandDArT_ReadyForGP_Dos.rds"))Markers Density
library(tidyverse); library(CMplot)
snps <- readRDS(file = here::here("output", "DCas22_6902", "DCas22_GBSandDArT_ReadyForGP_Dos.rds"))
CMsnps <- tibble(SNP = colnames(snps),
chr = substring(SNP,1,3),
pos = substring(SNP,4)) %>%
mutate(chr = gsub(pattern = "_", replacement = "", x = chr) %>%
gsub(pattern = "S", replacement = "") %>% as.integer,
pos = gsub(pattern = "_[A-Z]", replacement = "", x = pos) %>%
gsub(pattern = "_", replacement = "", x = .) %>% as.integer)
CMplot(CMsnps, plot.type = "d", bin.size = 1e6, col = c("darkgreen", "yellow", "red"),
file = "jpg", memo = "GBSDArTDensityMkrs", dpi = 500, file.output = T, verbose = TRUE)
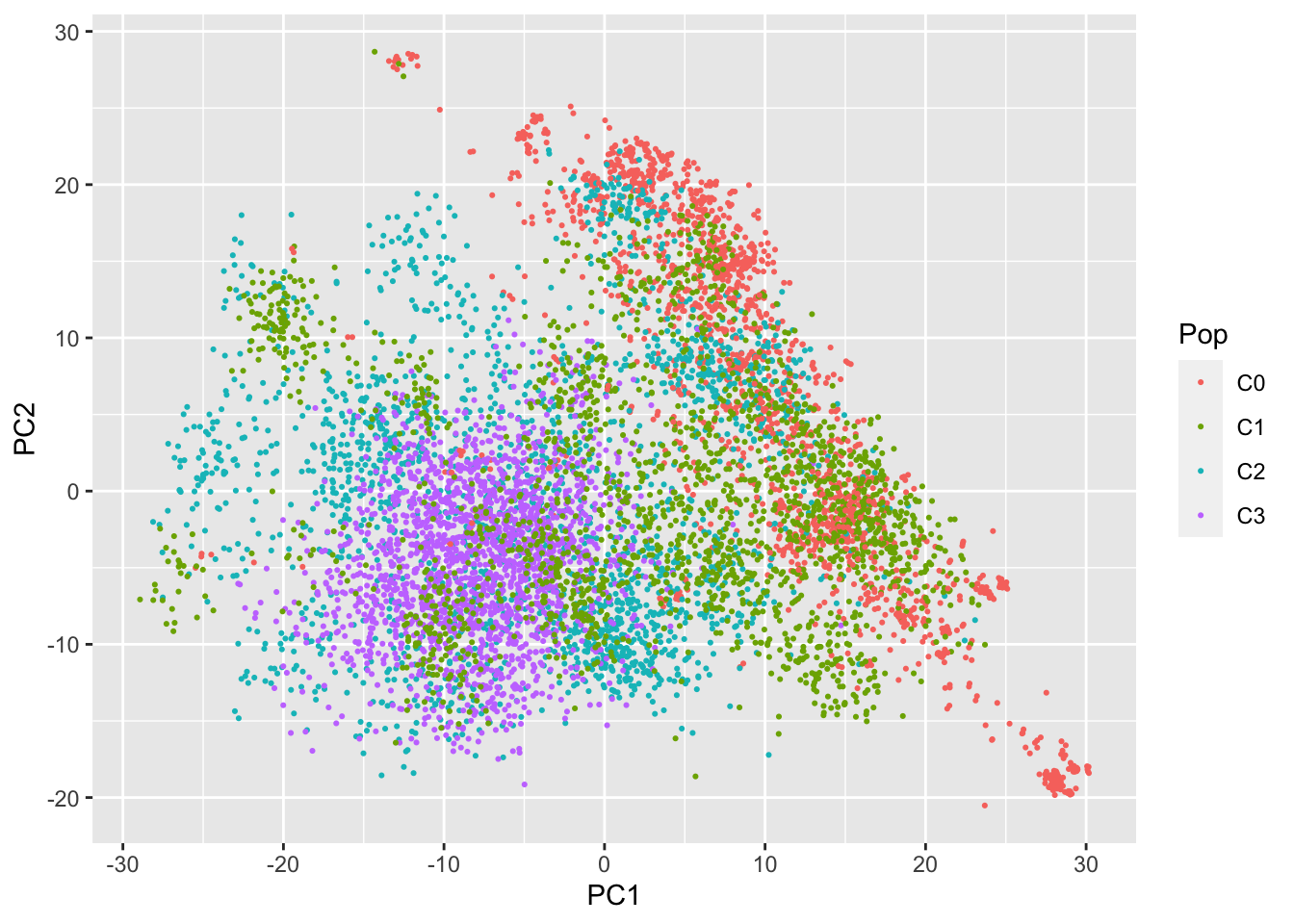
Principal Components Analysis
sessionInfo()R version 4.1.2 (2021-11-01)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Big Sur 11.6.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.1-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.1-arm64/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] data.table_1.14.3 reactable_0.2.3 magrittr_2.0.3 ggvenn_0.1.9
[5] forcats_0.5.1 stringr_1.4.0 dplyr_1.0.9 purrr_0.3.4
[9] readr_2.1.2 tidyr_1.2.0 tibble_3.1.7 ggplot2_3.3.6
[13] tidyverse_1.3.1 here_1.0.1
loaded via a namespace (and not attached):
[1] Rcpp_1.0.8.3 lubridate_1.8.0 assertthat_0.2.1 rprojroot_2.0.3
[5] digest_0.6.29 utf8_1.2.2 reactR_0.4.4 R6_2.5.1
[9] cellranger_1.1.0 backports_1.4.1 reprex_2.0.1 evaluate_0.15
[13] highr_0.9 httr_1.4.3 pillar_1.7.0 rlang_1.0.2
[17] readxl_1.4.0 rstudioapi_0.13 jquerylib_0.1.4 rmarkdown_2.14
[21] labeling_0.4.2 htmlwidgets_1.5.4 munsell_0.5.0 broom_0.8.0
[25] compiler_4.1.2 httpuv_1.6.5 modelr_0.1.8 xfun_0.30
[29] pkgconfig_2.0.3 htmltools_0.5.2 tidyselect_1.1.2 workflowr_1.7.0
[33] fansi_1.0.3 crayon_1.5.1 tzdb_0.3.0 dbplyr_2.1.1
[37] withr_2.5.0 later_1.3.0 jsonlite_1.8.0 gtable_0.3.0
[41] lifecycle_1.0.1 DBI_1.1.2 git2r_0.30.1 scales_1.2.0
[45] cli_3.3.0 stringi_1.7.6 farver_2.1.0 fs_1.5.2
[49] promises_1.2.0.1 xml2_1.3.3 bslib_0.3.1 ellipsis_0.3.2
[53] generics_0.1.2 vctrs_0.4.1 tools_4.1.2 glue_1.6.2
[57] crosstalk_1.2.0 hms_1.1.1 fastmap_1.1.0 yaml_2.3.5
[61] colorspace_2.0-3 rvest_1.0.2 knitr_1.38 haven_2.5.0
[65] sass_0.4.1